|
NanoStructures
1.0
DMFT solver for layered, strongly correlated nanostructures
|
Density matrix implementation of the numerical renormalization group. More...
#include <nrg.h>
Public Member Functions | |
| NRG (ChainProvider &chainProvider, Broadener &broadener) | |
| constructs an NRG algorithm instance The Wilson chain is provided by an implementation of the ChainProvider interface. The Broadener is used after diagonalization of the chain and calculation of the density matrix to broaden the representation of various correlation functions as a discrete set of delta peaks into continous functions. More... | |
| ~NRG () | |
| destructs the NRG algorithm object and frees all previously allocated memory. | |
| void | configure (config::Configuration &configuration) |
| configures the NRG instance with information in the configuration object. More... | |
| double | getU () const |
| retrieves the strength of the on-site impurity interaction U More... | |
| void | setU (double U) |
| sets the strength of the on-site impurity interaction U More... | |
| double | getEpsF () const |
| retrieves the on-site impurity energy \(\epsilon_f\) More... | |
| void | setEpsF (double epsF) |
| sets the on-site impurity energy \(\epsilon_f\) More... | |
| double | getTemperature () const |
| retrieves the system temperature T More... | |
| void | setTemperature (double T) |
| sets the system temperature T More... | |
| double | getClusterEnergy () const |
| returns the cluster energy. More... | |
| void | setClusterEnergy (double clusterEnergy) |
| sets the cluster energy to the indicated value. see getClusterEnergy(). More... | |
| double | getEnergyCutOff () const |
| returns the high energy cut-off for state truncation More... | |
| void | setEnergyCutOff (double energyCutOff) |
| sets the high energy cut-off for state truncation More... | |
| int | getMaxHilbertSpaceDimension () |
| returns the maximum allowed size of the HilberSpace. See truncateStates(int n). More... | |
| void | setMaxHilbertSpaceDimension (int maxHSdimension) |
| sets the maximum allowed size of the HilberSpace. See truncateStates(int n). More... | |
| int | getMaxIterations () |
| void | setMaxIterations (int maxIterations) |
| double | getOccupationUp () |
| returns the exp. value for spin- \(\uparrow\) electrons on the impurity. The actual calculation is performed in createPolesG_Up(int n). More... | |
| double | getOccupationDown () |
| returns the exp. value for spin- \(\downarrow\) electrons on the impurity. The actual calculation is performed in createPolesG_Down(int n). More... | |
| double | getOccupation () |
| returns the exp. value for electrons on the impurity. The actual calculation is performed in createPolesG_Up(int n) and createPolesG_Down(int n). More... | |
| double | getMagnetization () |
| returns the exp. value for the magnetization More... | |
| void | init () |
| prepares the NRG instance for the iterative diagonalization. More... | |
| void | setupInitialState () |
| initializes the impurity Hamiltonian \(H_0\). More... | |
| void | setupHamiltonian (int iteration) |
| iterates over all possible quantum number Q and Sz for iteration N and constructs the Hamiltonian for the corresponding HilbertSubSpace. More... | |
| void | truncateStates (int iteration) |
| marks all eigenstates above a cut-off energy \(E_c\) as discarded. More... | |
| void | propagateLocalMatrixElementUp (int iteration) |
| calculates the local impurity matrix elements or spin- \(\uparrow\) for a given iteration. More... | |
| void | propagateLocalMatrixElementDown (int iteration) |
| see propagateLocalMatrixElementUp(int iteration). This is the spin- \(\downarrow\) version. More... | |
| void | propagateLocalMatrixElementUp2 (int iteration) |
| calculates other local impurity matrix elements or spin- \(\uparrow\) for a given iteration. More... | |
| void | propagateLocalMatrixElementDown2 (int iteration) |
| see propagateLocalMatrixElementUp2(int iteration). This is the spin- \(\downarrow\) version. More... | |
| void | propagateChainOperatorElementsUp (int iteration) |
| calculates the chain matrix elements for spin- \(\uparrow\) for a given iteration. More... | |
| void | propagateChainOperatorElementsDown (int iteration) |
| see propagateChainOperatorElementsUp(int iteration). This is the spin- \(\downarrow\) version. More... | |
| void | createPolesG_Up (int iteration) |
| calculates delta peaks for the spin- \(\uparrow\) impurity Green's function. More... | |
| void | createPolesG_Down (int iteration) |
| see createPolesG_Up(int iteration). This is the spin- \(\downarrow\) version. More... | |
| void | createPolesF_Up (int iteration) |
| calculates delta peaks for the spin- \(\uparrow\) impurity correlator used in the self-energy trick. More... | |
| void | createPolesF_Down (int iteration) |
| see createPolesF_Down(int iteration). This is the spin- \(\downarrow\) version. More... | |
| void | solve (bool silent=false) |
| solves the impurity problem. More... | |
| void | solve_symmetric_SZ (bool silent=false) |
| Version of solve(bool silent) for a Wilson chain with Sz symmetry. More... | |
| void | builDM () |
| performs a backward run to construct the reduced density matrices | |
| void | showInfo () |
| lists the physical and numerical parameters for the NRG instance. Also dumps info about the Broadener object. | |
| void | deleteChainOperatorElements (int iteration) |
| deletes all chain operator matrix elements for the indicated iteration. More... | |
| void | deleteTransformationMatrices (int iteration) |
| deletes all transformation matrices for the indicated iteration. More... | |
| void | deleteDensityMatrices (int iteration) |
| deletes all chain operator matrix elements for the indicated iteration. More... | |
| void | deleteImpurityMatrixElements (int iteration) |
| deletes all impurity operator matrix elements for the indicated iteration. More... | |
| void | getSelfEnergy (math::CFunction &SUp, math::CFunction &SDown) |
| returns the impurity selfenergy. More... | |
| void | getSelfEnergy (math::CFunction &S) |
| returns the impurity selfenergy. More... | |
| void | getGreensFunction (math::CFunction &GUp, math::CFunction &GDown) |
| returns the impurity Green's function. More... | |
| void | getGreensFunction (math::CFunction &G) |
| returns the Green's function. More... | |
| void | getFFunction (math::CFunction &FUp, math::CFunction &FDown) |
| returns the correlator for the self-energy trick More... | |
| void | getFFunction (math::CFunction &F) |
| returns the correlator for the self-energy trick More... | |
Protected Attributes | |
| ChainProvider & | m_chainProvider |
| Broadener & | m_broadener |
| Broadener * | m_G_Up |
| Broadener * | m_G_Down |
| Broadener * | m_F_Up |
| Broadener * | m_F_Down |
| math::CFunction | m_F_S_Up |
| math::CFunction | m_F_S_Down |
| math::CFunction | m_F_G_Up |
| math::CFunction | m_F_G_Down |
| math::CFunction | m_F_F_Up |
| math::CFunction | m_F_F_Down |
| double | m_n_Up |
| double | m_n_Down |
| double | m_epsF |
| double | m_U |
| double | m_temperature |
| int | m_maxIterations |
| std::mathbfor< double > | m_energies |
| HilbertSpaceTable | m_hilbertSpaces |
| double | m_clusterEnergy |
| double | m_energyCutOff |
| int | m_maxHSdimension |
| int | m_nFirstTruncated |
| int | signLME [4] |
| int | dQ [4] |
| int | dSz [4] |
Detailed Description
Density matrix implementation of the numerical renormalization group.
The Numerical Renormalization Group
The Numerical Renormalization Group (NRG) is one of the standard tools to study correlation effects in quantum impurity models. Here a small interacting subsystem with a small number of degrees of freedom (the impurity) is coupled to a bath of fermions. No restriction exists as to the structure of the impurity subsystem. The bath however must consist of non-interacting fermions.
Logarithmic discretization
At the heart of the NRG lies a logarithmic discretization of the continous conduction electron band. The continuum density of states \(\rho(\epsilon)\) is approximated by a discrete set of delta poles. By introducing a discretisation parameter \(\Lambda\) Wilson divided the normalised energy range \([-1,1]\) into \(2n\) intervals where the \(n\)th interval (for positive \(\epsilon\)) extends from \(\Lambda^{-(n+1)}\) to \(\Lambda^n\). The logarithmic discretisation separates the electron energies into different orders of magnitude where energies close to the Fermi level \(k_B T \ll D\) with \(D\) the bandwidth, which determine the low temperature properties, are well sampled.
Mapping onto a chain
In order to solve the discretized Hamiltonian iteratively one introduces a set of operators \(f_{n\sigma}\) with \(n>0\) in such a way that they exhibit only nearest neighbour coupling. The Hamiltonian assumes the structure of a hopping Hamiltonian on a semi-infinite chain, which is often referred to as the Wilson chain.
Iterative diagonalization
The transformations performed so far have rendered a form of the Hamiltonian which is amendable to an iterative diagonalisation procedure. Now one defines a sequence of Hamiltonians \(H_N\) with \(N \geq 0\). The full discrete Hamiltonian is recovered in the limit \(N\to\infty\) as
\( \begin{equation} H = \lim_{N\to\infty} \frac{1}{2} (1+\Lambda^{-1}) D \Lambda^{-(N-1)/2}H_N \end{equation} \)
Two successive Hamiltonians in the series are connected by the recursion relation
\( \begin{equation} H_{N+1} = \Lambda^{1/2} H_N + t_N ( f^\dagger_{N \sigma} f_{N+1\sigma} + f^\dagger _{N+1\sigma} f_{N\sigma} ) \end{equation} \)
with the initial Hamiltonian in the series containing the impurity itself given by
\( \begin{equation} H_0 = \Lambda^{-\frac{1}{2}} \left[ \tilde{\delta_d} c^\dagger_{d \sigma} c_{d \sigma} + \tilde{\Gamma}^{1/2} ( f^\dagger_{0 \sigma} c_{d\sigma} + \text{h.c.} ) + \tilde{U} ( c_{d \sigma}^\dagger c_{d \sigma} - 1)^2\right] \end{equation} \)
In this form the single impurity Anderson model can be efficiently solved on a computer by taking advantage of the renormalisation group character of the above description. One starts with a diagonalisation of \(H_0\) which can be easily accomplished numerically. Assuming that we have diagonalised a Wilson chain of length \(m\) and that the eigenstates are given by \(|\mathbf r;m\rangle\) we construct a product basis for the Wilson chain of length \(m+1\) by
\( \begin{equation} |(\mathbf r,\alpha_{m+1});m+1 \rangle = |\mathbf r; m \rangle \otimes |\alpha_{m+1}\rangle \end{equation} \)
where \(|\alpha_{m+1}\rangle\) are the eigenstates of the decoupled site \(|\alpha_{m+1}\rangle=\{ |\rangle,|\uparrow\rangle,|\downarrow\rangle,| \uparrow \downarrow \rangle \}\). The matrix elements of the Hamiltonian for the Wilson chain of length \(m+1\) for this product basis are given by
\( \begin{align} \langle(\mathbf r',\alpha'_{m+1}); m+1|&H_{m+1} |(\mathbf r,\alpha_{m+1}); m+1\rangle = \Lambda^{1/2} E_{\mathbf r,m} \delta_{\mathbf r \mathbf r'} \delta_{\alpha \alpha'} \nonumber \\ &+\left( \langle \mathbf r'; m |f^\dagger_{m\sigma} |\mathbf r;m \rangle \langle \alpha' | f_{m+1\alpha} |\alpha \rangle + \langle \mathbf r'; m | f_{m\sigma} |\mathbf r;m \rangle \langle\alpha'| f^\dagger_{m+1\alpha} |\alpha\rangle\right) \end{align} \)
The eigenvalue problem for the chain of length \(m+1\) can therefore be solved numerically using only a knowledge of the eigenspectrum of the chain of length \(m\) and the matrix elements of the operators \(f_{m\sigma}^\dagger\). Diagonalising the Hamiltonian \(H_{m+1}\), set up in the above product basis, can be described by a unitary transformation
\( \begin{equation} |\mathbf r'; m+1 \rangle = \sum_{\alpha_{m+1}, \mathbf r} U^{\alpha_{m+1}}_{\mathbf r', \mathbf r} |\mathbf r;m\rangle \otimes |\alpha_{m+1}\rangle \end{equation} \)
where \(|\mathbf r'; m+1 \rangle\) denotes the new eigenbasis of the Hamiltonian \(H_{m+1}\).
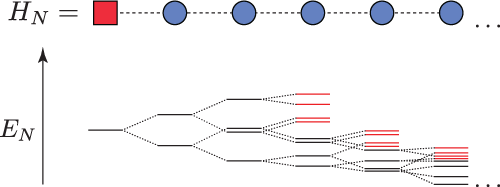
iterative diagonalisation splits each energy level into 4 levels upon the addition of another chain element. In this schematic picture however each energy level is split into only two levels in order not to make the illustration to cluttered. Due to the exponential decrease in the couplings it is save to truncate the high energy states without altering the spectrum of the low energy states. The truncated states are marked red."
References
Constructor & Destructor Documentation
| nrg::NRG::NRG | ( | ChainProvider & | chainProvider, |
| Broadener & | broadener | ||
| ) |
constructs an NRG algorithm instance The Wilson chain is provided by an implementation of the ChainProvider interface. The Broadener is used after diagonalization of the chain and calculation of the density matrix to broaden the representation of various correlation functions as a discrete set of delta peaks into continous functions.
- Parameters
-
[in] chainProvider implementation of the ChainProvider interface; supplies the Wilson chain. [in] broadener implementation of the Broadener interface; broadens the discrete set of delta peaks of the correlation functions into continous functions.
Member Function Documentation
| void nrg::NRG::configure | ( | config::Configuration & | configuration | ) |
configures the NRG instance with information in the configuration object.
- Parameters
-
[in] configuration a configuration object which can be queried for various numerical and physical parameters concerning the impurity problem.
| void nrg::NRG::createPolesF_Down | ( | int | iteration | ) |
see createPolesF_Down(int iteration). This is the spin- \(\downarrow\) version.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::createPolesF_Up | ( | int | iteration | ) |
calculates delta peaks for the spin- \(\uparrow\) impurity correlator used in the self-energy trick.
This function uses the reduced density matrix and the impurity operator matrix elements to calculate delta peaks of the Lehmann representation of the spin- \(\uparrow\) correlator for the given iteration used in the self-energy trick.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::createPolesG_Down | ( | int | iteration | ) |
see createPolesG_Up(int iteration). This is the spin- \(\downarrow\) version.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::createPolesG_Up | ( | int | iteration | ) |
calculates delta peaks for the spin- \(\uparrow\) impurity Green's function.
This function uses the reduced density matrix and the impurity operator matrix elements to calculate delta peaks of the Lehmann representation of the spin- \(\uparrow\) impurity Green's function for the given iteration.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::deleteChainOperatorElements | ( | int | iteration | ) |
deletes all chain operator matrix elements for the indicated iteration.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::deleteDensityMatrices | ( | int | iteration | ) |
deletes all chain operator matrix elements for the indicated iteration.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::deleteImpurityMatrixElements | ( | int | iteration | ) |
deletes all impurity operator matrix elements for the indicated iteration.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::deleteTransformationMatrices | ( | int | iteration | ) |
deletes all transformation matrices for the indicated iteration.
- Parameters
-
[in] iteration NRG iteration
|
inline |
returns the cluster energy.
Symmetries create degeneracies in the spectrum of eigenstates. Due to numerical noise these states can energetically drift apart by tiny amounts. It is generally not a good idea to cut-off within a multiplet (or "cluster"). It is ensured that no cut is made in between two energy levels less than a certain energy difference apart. This function returns this energy difference.
- Returns
- cluster energy
|
inline |
returns the high energy cut-off for state truncation
- Returns
- high-energy cut-off
|
inline |
retrieves the on-site impurity energy \(\epsilon_f\)
- Returns
- epsF on-site energy \(\epsilon_f\)
|
inline |
returns the correlator for the self-energy trick
- Parameters
-
FUp reference to spin- \(\uparrow\) correlator FDown reference to spin- \(\downarrow\) correlator
|
inline |
returns the correlator for the self-energy trick
- Parameters
-
F reference to spin- \(\uparrow\) correlator
|
inline |
returns the impurity Green's function.
- Parameters
-
[out] GUp reference to spin- \(\uparrow\) Green's function [out] GDown reference to spin- \(\downarrow\) Green's function
|
inline |
returns the Green's function.
- Parameters
-
[out] G reference to spin- \(\uparrow\) Green's function
|
inline |
returns the exp. value for the magnetization
The actual calculation is performed in createPolesG_Up(int n) and createPolesG_Down(int n). The magnetization is calculated as \( \avg{m} = \avg{n_\uparrow} - \avg{\downarrow}\)
- Returns
|
inline |
returns the maximum allowed size of the HilberSpace. See truncateStates(int n).
- Returns
- max dimension of HilbertSpace
|
inline |
|
inline |
returns the exp. value for electrons on the impurity. The actual calculation is performed in createPolesG_Up(int n) and createPolesG_Down(int n).
- Returns
|
inline |
returns the exp. value for spin- \(\downarrow\) electrons on the impurity. The actual calculation is performed in createPolesG_Down(int n).
- Returns
|
inline |
returns the exp. value for spin- \(\uparrow\) electrons on the impurity. The actual calculation is performed in createPolesG_Up(int n).
- Returns
|
inline |
returns the impurity selfenergy.
- Parameters
-
[out] SUp reference to spin- \(\uparrow\) self energy [out] SDown reference to spin- \(\downarrow\) self energy
|
inline |
returns the impurity selfenergy.
- Parameters
-
[out] S reference to spin- \(\uparrow\) self energy
|
inline |
retrieves the system temperature T
- Returns
- system temperature T
|
inline |
retrieves the strength of the on-site impurity interaction U
- Returns
- on-site impurity interaction U
| void nrg::NRG::init | ( | ) |
prepares the NRG instance for the iterative diagonalization.
The function performs the following steps:
Initialization
- If the max. iterations have not been set, chooses them automatically based on the system temperature T.
- Checks whether enough NRG iteration will be performed for the given temperature T.
- Calls the ChainProvider to prepare a chain of the appropriate length,
- Sets the temperature T in the broadeners and initializes them.
- Calls setupInitialState() to initialize the impurity Hamiltonian \(H_0\).
| void nrg::NRG::propagateChainOperatorElementsDown | ( | int | iteration | ) |
see propagateChainOperatorElementsUp(int iteration). This is the spin- \(\downarrow\) version.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::propagateChainOperatorElementsUp | ( | int | iteration | ) |
calculates the chain matrix elements for spin- \(\uparrow\) for a given iteration.
Assuming that we have diagonalised a Wilson chain of length \(m\) and that the eigenstates are given by \(|\mathbf r;m\rangle\) we construct a product basis for the Wilson chain of length \(m+1\) by
\( \begin{equation} |(\mathbf r,\alpha_{m+1});m+1\rangle = |\mathbf r; m\rangle \otimes |\alpha_{m+1}\rangle \end{equation} \)
where \(|\alpha_{m+1}\rangle\) are the eigenstates of the decoupled site \(|\alpha_{m+1}\rangle=\{ |\rangle,|\uparrow\rangle,|\downarrow\rangle,| \uparrow \downarrow \rangle \}\). The matrix elements of the Hamiltonian for the Wilson chain of length \(m+1\) for this product basis are given by
\( \begin{align} \langle(\mathbf r',\alpha'_{m+1}); m+1| &H_{m+1} |(\mathbf r,\alpha_{m+1}); m+1\rangle = \Lambda^{1/2} E_{\mathbf r,m} \delta_{\mathbf r \mathbf r'} \delta_{\alpha \alpha'} \nonumber \\ &+\left( \langle \mathbf r'; m | f^\dagger_{m\sigma} | \mathbf r;m \rangle \langle \alpha'| f_{m+1\alpha} |\alpha \rangle + \langle \mathbf r'; m | f_{m\sigma} |\mathbf r;m\rangle \langle\alpha'| f^\dagger_{m+1\alpha} |\alpha\rangle\right). \end{align} \)
It is precisely the matrix elements \( \langle \mathbf r'; m | f^\dagger_{m\uparrow} | \mathbf r;m \rangle \), which are calculated in this function.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::propagateLocalMatrixElementDown | ( | int | iteration | ) |
see propagateLocalMatrixElementUp(int iteration). This is the spin- \(\downarrow\) version.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::propagateLocalMatrixElementDown2 | ( | int | iteration | ) |
see propagateLocalMatrixElementUp2(int iteration). This is the spin- \(\downarrow\) version.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::propagateLocalMatrixElementUp | ( | int | iteration | ) |
calculates the local impurity matrix elements or spin- \(\uparrow\) for a given iteration.
Lehmann resolving the impurity spectral function one obtains
\( \begin{equation} A_\sigma(\omega) = \sum_{a,b} \bra{b} c_{d\sigma}\ket{a} \frac{\exp{\left[ -\beta E_a \right]}}{Z} \bra{a}c_{d\sigma}^\dagger\ket{b} \delta(\omega + E_a - E_b) \end{equation} \)
where $Z$ is the total partition function $Z = {[ - E_a ]}$, ${a}$ and ${b}$ are a complete set of states and $E_a$ is the eigenenergy of state ${a}$. We see that the Lehmann representation gathers the necessary information to construct the spectrum from knowledge of certain matrix elements encoding hopping processes between the impurity and the conduction electron band. It is precisely these matrix elements \( \langle \mathbf r'; m | c^\dagger_{\uparrow} | \mathbf r;m \rangle \delta_{\alpha' \alpha} \) which are calculated in this function.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::propagateLocalMatrixElementUp2 | ( | int | iteration | ) |
calculates other local impurity matrix elements or spin- \(\uparrow\) for a given iteration.
Bulla et al. first showed that it is possible to write the self-energy as the ratio of two correlation functions, both of which can be calculated directly within the NRG. An equation of motion technique is used to show that the self-energy is given by,
\( \begin{equation} \Sigma_\sigma(\omega) = U \frac{F_{\sigma}(\omega)}{G_{\sigma}(\omega)} \end{equation} \)
where $G_{}()$ is the impurity Green's function defined as
\( \begin{equation} G_{\sigma}(\omega) = -i \int_{-\infty}^\infty dt~e^{ i \omega t } \Theta(t) \avg{ \left\{ f_{\sigma}(t), f_{\sigma}^\dagger \right\} } \end{equation} \)
and $F_{}()$ is an auxiliary correlation function given by
\( \begin{equation} F_{\sigma}(\omega) = -i \int_{-\infty}^\infty dt e^{ i \omega t } \Theta(t) \avg{ \left\{ \left( f_{\bar{\sigma}}^\dagger f_{\bar{\sigma}} f_{\sigma}\right)(t), f_{\sigma}^\dagger \right\} } \end{equation} \)
The imaginary parts of $F_{}()$ and $G_{}()$ are calculated from NRG data using the Lehman sum within the full density matrix approach, with the poles of the spectrum broadened as above. The real parts are then obtained by Kramers-Kronig transform. Discretization artifacts cancel to some extent by dividing the two quantities. This produces a rather smooth self-energy, which in term can be used to calculate an improved spectrum for the impurity. Z-averaging can also be used to further increase accuracy and resolution.
It is precisely these matrix elements \( \langle \mathbf r'; m | c^\dagger_{\downarrow} c^\phantom{\dagger}_{\downarrow} c^\dagger_{\uparrow} | \mathbf r;m \rangle \delta_{\alpha' \alpha} \) which are calculated in this function.
- Parameters
-
[in] iteration NRG iteration
|
inline |
sets the cluster energy to the indicated value. see getClusterEnergy().
- Parameters
-
clusterEnergy
|
inline |
sets the high energy cut-off for state truncation
- Parameters
-
[in] clusterEnergy new high-energy cut-off
|
inline |
sets the on-site impurity energy \(\epsilon_f\)
- Parameters
-
[in] epsF on-site energy \(\epsilon_f\)
|
inline |
sets the maximum allowed size of the HilberSpace. See truncateStates(int n).
It is advisable to set this number to a large value (so that it does not get in the way) and let the energy cut-off( see setEnergyCutOff(double energyCutOff) ) do the truncation.
- Parameters
-
[in] maxHSdimension max. dimension of the HilbertSpace
|
inline |
|
inline |
sets the system temperature T
- Parameters
-
[in] T system temperature T
|
inline |
sets the strength of the on-site impurity interaction U
- Parameters
-
U on-site impurity interaction U
| void nrg::NRG::setupHamiltonian | ( | int | iteration | ) |
iterates over all possible quantum number Q and Sz for iteration N and constructs the Hamiltonian for the corresponding HilbertSubSpace.
For a given HilbertSubSpace (HSS) in iteration N the function looks up the appropriate HSSs (up to 4) of iteration N-1 and uses the chain matrix elements to construct the Hamiltonian for the HSS in iteration N. Then the Hamiltonian is diagonalized using a call to Lapack's dsyev Lapack replaces the Hamiltonian m_H in the HSS by the unitary transformation that diagonalized it and places and sorts the eigenvalues (energies) into the HSS object as well. The function discards the chain matrix elements of iteration N-1, which are no longer needed.
- Parameters
-
[in] iteration NRG iteration
| void nrg::NRG::setupInitialState | ( | ) |
initializes the impurity Hamiltonian \(H_0\).
This function creates the first 4 HilbertSubSpace instances for the chain consisting only of the impurity ( \( \{ |\rangle,|\uparrow\rangle,|\downarrow\rangle,| \uparrow \downarrow \rangle \}\) ). It determines the eigenenergies and both the local impurity and the chain matrix elements.
| void nrg::NRG::solve | ( | bool | silent = false | ) |
solves the impurity problem.
First, the function queries the ChainProvider whether the provided chain is symmetric with respect to the Sz quantum number. If so, it suffices to calculate all matrix elements and correlator for spin- \(\uparrow\) only.
The algorithm moves along the Wilson chain iteratively in a forward-backward- forward pattern.
Step 1 - Forward run: Iterative diagonalization
In the first forward run, the Hamiltonian is iteratively constructed and diagonalized (using setupHamiltonian(int n) ). Chain matrix elements are calculated along the way as needed. Once the highest eigenenergy of the chain exceeds the predefined threshold value, the truncation sets in (see truncateStates(int iteration) ) and only the kept states are used to construct the HilbertSpace of the next iteration.
Step 2 - Backward run: Reduced density matrix
In a second step, buildDM() is called, which performs a backward run starting from the last iteration and calculates the reduced density matrix for the different iterations.
Step 3 - Forward run: Calculation of Correlators
In a third step, a forward run is performed which uses the reduced density matrix to calculate impurity matrix elements. The matrices are propagated from iteration to iteration and used to calculate an approximate Lehmann representation of the impurity Green's function and a another correlation function which is needed for the self-energy trick.
Finally, the discrete set of delta peaks for each correlator is broadened to get continous functions (see Broadener).
- Parameters
-
[in] silent if true, silences the impurity solver (useful for DMFT)
| void nrg::NRG::solve_symmetric_SZ | ( | bool | silent = false | ) |
Version of solve(bool silent) for a Wilson chain with Sz symmetry.
- Parameters
-
silent if true, silences the impurity solver (useful for DMFT)
| void nrg::NRG::truncateStates | ( | int | iteration | ) |
marks all eigenstates above a cut-off energy \(E_c\) as discarded.
The Hilbertspace grows exponentially with the length of the Wilson chain. Therefore the eigenstates have to be truncated above an energy cutoff (see void setEnergyCutOff(double energyCutOff) ). Only the "kept" states in iteration N-1 with an energy smaller than the cut-off are used to construct the Hilbertspace for iteration N. A separation of energy scales is achieved by the logarithmic discretization of the energy band and justifies the truncation: States with a high energy in iteration N-1 cannot affect the structure of the low-energy states in iteration N.
Special attention has to be paid to the role of symmetries. Symmetries create degeneracies in the spectrum of eigenstates. Due to numerical noise these states can energetically drift apart by tiny amounts. It is generally not a good idea to cut-off within a multiplet. It is ensured that no cut is made in between two energy levels less than a certain energy difference apart (see void setClusterEnergy(double clusterEnergy) ).
- Parameters
-
[in] iteration NRG iteration
The documentation for this class was generated from the following files:
- /home/chris/Development/Release/NanoStructures/nrg/nrg.h
- /home/chris/Development/Release/NanoStructures/nrg/nrg.cpp
Generated on Sat Jun 28 2014 11:55:18 for NanoStructures by
 1.8.6
1.8.6